

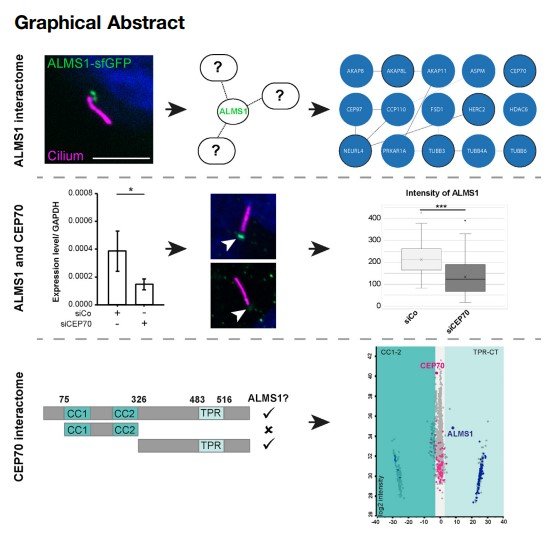
Alström syndrome (ALMS) is a very rare autosomalrecessive disorder, causing a broad range of clinical defects most notably retinal degeneration, type 2 diabetes, and truncal obesity. The ALMS1 gene encodes a complex and huge ~0.5 MDa protein, which has hampered analysis in the past. The ALMS1 protein is localized to the centrioles and the basal body of cilia and is involved in signaling processes, for example, TGF-β signaling. However, the exact molecular function of ALMS1 at the basal body remains elusive and controversial. We recently demonstrated that protein complex analysis utilizing endogenously tagged cells provides an excellent tool to investigate protein interactions of ciliary proteins. Here, CRISPR/Cas9- mediated endogenously tagged ALMS1 cells were used for affinity-based protein complex analysis. Centrosomal and microtubule-associated proteins were identified, which are potential regulators of ALMS1 function, such as the centrosomal protein 70 kDa (CEP70). Candidate proteins were further investigated in ALMS1-deficient hTERTRPE1 cells. Loss of ALMS1 led to shortened cilia with no change in structural protein localization, for example, acetylated and ɣ-tubulin, Centrin-3, or the novel interactor CEP70. Conversely, reduction of CEP70 resulted in decreased ALMS1 at the ciliary basal body. Complex analysis of CEP70 revealed domain-specific ALMS1 interaction involving the TPR-containing C-terminal (TRP-CT) fragment of CEP70. In addition to ALMS1, several ciliary proteins, including CEP135, were found to specifically bind to the TPR-CT domain. Data are available via ProteomeXchange with the identifier PXD046401. Protein interactors identified in this study provide candidate lists that help to understand ALMS1 and CEP70 function in ciliarelated protein modification, cell death, and diseaserelated mechanisms.